Tổng quan
Hội chứng Smith-Lemli-Opitz là gì?
Hội chứng Smith-Lemli-Opitz là một rối loạn phát triển đặc trưng bởi đặc tính khác biệt trên khuôn mặt, kích thước đầu nhỏ, các vấn đề khuyết tật về học tập và trí tuệ, các vấn đề hành vi. Các dị tật ở tim, phổi, thận, đường tiêu hóa và cơ quan sinh dục cũng có thể xảy ra.
Mức độ phổ biến của hội chứng Smith-Lemli-Opitz
Hội chứng Smith-Lemli-Opitz rất hiếm gặp. Hãy thảo luận với bác sĩ để biết thêm thông tin.
Triệu chứng
Những dấu hiệu và triệu chứng của Hội chứng Smith-Lemli-Opitz là gì?
Các triệu chứng phổ biến của hội chứng Smith-Lemli-Opitz là:
Chậm phát triển
Chậm phát triển Tâm thần vận động
Chậm phát triển tâm thần
Các dị tật cơ xương bao gồm dư ngón tay hoặc ngón chân hoặc dính các ngón tay hoặc các ngón chân
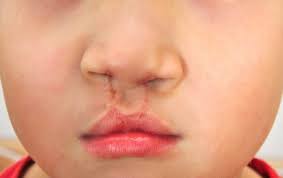
Hở hàm ếch
Bộ phận sinh dục ngoài của nam giới kém phát triển
Sụp mí mắt
Các nếp nhăn ở mí Mắt trên và dưới
Hàm nhỏ
Tai lớn
Các vấn đề về thị lực dưới hình thức đục thủy tinh thể
Các vấn đề Thần kinh như co giật
Giảm trương lực
Tắc nghẽn ruột
Nhạy cảm ánh sáng
Bạn có thể gặp các triệu chứng khác không được đề cập. Nếu bạn có bất kỳ thắc mắc nào về các dấu hiệu bệnh, hãy tham khảo ý kiến bác sĩ.
Khi nào bạn cần gặp bác sĩ?
Nếu bạn có bất kỳ dấu hiệu hoặc triệu chứng nêu trên hoặc có bất kỳ câu hỏi nào, xin vui lòng tham khảo ý kiến bác sĩ. Cơ địa mỗi người là khác nhau, vì vậy hãy hỏi ý kiến bác sĩ để lựa chọn được phương án thích hợp nhất.
Nguyên nhân
Nguyên nhân nào gây ra hội chứng Smith-Lemli-Opitz?
Nguyên nhân chính gây hội chứng Smith-Lemli-Opitz là do đột biến gen DHCR7 dẫn đến sự thiếu hụt enzyme reductase 7-dehydrocholesterol. Chức năng của enzyme này là thúc đẩy quá trình chuyển hóa cholesterol.
Hội chứng Smith-Lemli-Opitz theo mô hình nhiễm sắc thể thường mang tính trạng lặn, nghĩa là cần có hai bản sao của gen bị lỗi từ bố và mẹ để phát triển hội chứng Smith-Lemli-Opitz ở trẻ.
Nếu một cá nhân chỉ nhận được một gen khiếm khuyết thì người đó sẽ là người chuyên chở gen bệnh và không hiển thị bất kỳ triệu chứng nào của hội chứng Smith-Lemli-Opitz.
Triệu chứng
Chẩn đoán
Điều trị
Click vào ảnh để xem 3 hình ảnh minh họa
Những thông tin được cung cấp không thể thay thế cho lời khuyên của các chuyên viên y tế. Hãy luôn tham khảo ý kiến bác sĩ.
Những kỹ thuật y tế nào dùng để chẩn đoán hội chứng Smith-Lemli-Opitz?
Để chẩn đoán hội chứng Smith-Lemli-Opitz, đầu tiên bác sĩ sẽ thu thập bệnh sử chi tiết của bệnh nhân bao gồm cả lịch sử gia đình để xác định xem có bất kỳ thành viên nào khác trong gia đình có những triệu chứng tương tự.
Khi nghi ngờ hội chứng Smith-Lemli-Opitz, xét nghiệm enzyme sẽ được thực hiện để phát hiện sự thiếu hụt enzyme 7-dehydrocholesterol reductase.
Xét nghiệm gen có thể được thực hiện để tìm ra đột biến gen DHCR7.
Những phương pháp nào dùng để điều trị hội chứng Smith-Lemli-Opitz?
Không có điều trị đặc hiệu cho hội chứng và điều trị hội chứng Smith-Lemli-Opitz chủ yếu nhằm cải thiện triệu chứng và sửa chữa các khiếm khuyết phát sinh từ rối loạn này.
Bệnh nhân sẽ được đánh giá chặt chẽ bởi một nhóm các chuyên gia phối hợp hầu như tất cả các chuyên ngành. Bệnh nhân sẽ được theo dõi các khiếm khuyết ở tim, mắt, thần kinh, thần kinh cơ, tiêu hóa, tiết niệu sinh dục và lên một kế hoạch điều trị phù hợp.
Phẫu thuật có thể được chỉ định để sửa chữa sứt môi hoặc dính các ngón chân và các ngón tay. Một số dị tật tim cũng có thể được điều trị bằng phẫu thuật. Các bất thường về bộ phận sinh dục ở nam giới mắc hội chứng Smith-Lemli-Opitz cũng cần phải được phẫu thuật.
Trong một số trường hợp, bổ sung cholesterol có thể được bác sĩ khuyến khích để nâng cao tốc độ tăng trưởng cũng như tình trạng chung của bệnh nhân.